Imagine a patient diagnosed with a rare, aggressive form of leukemia. For decades, the prognosis was a terminal countdown, but today, they receive a targeted infusion that puts the disease into complete remission within months.
While we rightly celebrate the pharmaceutical breakthrough and the clinicians at the bedside, we often overlook the invisible structural framework that made this miracle possible: the protocol. Every life-saving treatment begins its journey not just in a laboratory, but within a meticulously drafted document that maps every heartbeat, blood draw, and data point of the clinical process.
This article aims to demystify the anatomy of a clinical trial protocol, peeling back the technical layers to reveal how scientific rigor is converted into medical progress. At ScientistsHub, we often ask: How do we ensure that a medical breakthrough is not just a stroke of luck, but a repeatable, verifiable victory for human health? The answer is found in the design. To truly appreciate the result of any trial, one must first master the architecture of the inquiry.
The Architecture of Inquiry: What is a Clinical Protocol?
A clinical protocol is far more than a simple set of instructions; it is a strategic mandate that balances the boundaries of scientific curiosity with the rigid, non-negotiable standards of human safety. It functions simultaneously as a "scientific recipe" for discovery and a "legal contract" between the researchers, the participants, and regulatory bodies (FDA, 2022).

Upon its first introduction into the trial ecosystem, the Protocol serves as the definitive authority on how the study will be conducted, ensuring that the trial's execution is shielded from bias and operational drift.
Every blueprint is designed to satisfy three primary objectives (ICH, 2016):
Patient Safety: Ensuring that the risks to participants are minimized and ethically justified by the potential benefits.
Data Integrity: Guaranteeing that the information collected is accurate, complete, and verifiable for regulatory review.
Reproducibility: Providing enough detail so that another scientific team can replicate the study and achieve the same results, confirming the findings are not an anomaly.
Once these objectives are established, the document moves from abstract goals to specific structural components that dictate the trial’s population and goals.
The Blueprint’s Bones: Inclusion, Exclusion, and Endpoints
The success or failure of a trial is often decided before the first patient is even enrolled, dictated by the "Inclusion and Exclusion Criteria." These parameters define the specific population that will participate in the study. From a scientific perspective, narrow criteria are preferred because they increase the "signal-to-noise" ratio, allowing researchers to see the drug’s effect clearly without the interference of outside health variables.
However, this creates a significant strategic tension. If the criteria are too narrow, it leads to an "enrollment bottleneck," slowing down the trial for years and resulting in data that may fail to predict how a drug behaves in the "messy" reality of the real world, particularly in geriatric or multi-morbid populations who were excluded from the study (FDA, 2022).
To measure success, the protocol establishes a hierarchy of goals known as endpoints:
Primary Endpoints: These are the critical outcomes—the "must-haves"—that determine if a trial has met its main objective, such as a statistically significant reduction in mortality or tumor size.
Secondary Endpoints: These provide supportive data, such as the drug's impact on a patient's quality of life or secondary symptoms, adding nuance to the primary findings.
While the science behind these endpoints must be sound, the ethical framework surrounding the participants must be impenetrable.
The Ethical Scaffolding: Safeguarding the Human Element
The protocol acts as an ethical shield, ensuring that human subjects are never treated as mere data points. No trial can proceed without the oversight of Institutional Review Boards (IRBs), which vet the protocol to ensure it upholds the mandate of Informed Consent (World Medical Association, 2013). This process ensures that every participant is fully aware of the risks and benefits before agreeing to take part.
Furthermore, the protocol mandates the involvement of Data Monitoring Committees (DMC). These independent groups provide real-time oversight, maintaining the absolute necessity of participant welfare over scientific gain (ICH, 2016). If the data suggests a safety concern, the DMC has the power to stop the trial immediately to protect participants.
A protocol is a sacred document. In clinical investigation, 'protocol drift' is the silent killer of validity. We rarely modify the blueprint mid-stream because any significant amendment risks compromising the 'Statistical Power' of the study. Moving the goalposts to favor a result isn't just bad science; it's a regulatory violation.
This rigidity is the mechanism that produces the "certain" data required to change the course of medicine.
Data & Evidence: The Statistics of Certainty
The arbiter of truth within any protocol is the Statistical Analysis Plan (SAP). This plan effectively "locks the vault" on the data before the first patient is even unblinded (ICH, 2016). By pre-defining exactly how the data will be analyzed, the SAP prevents "p-hacking" or data dredging—the biased practice of searching through data until a statistically significant pattern emerges by pure chance.
Many trials fail at this stage because of flawed protocol design, such as unrealistic recruitment targets that leave the study "underpowered." To understand if a trial is successful, we look at p-values (the probability that the observed result happened by chance, with a value typically below 0.05 signaling success) and confidence intervals (the range in which the true effect of the treatment likely falls). These metrics, governed by the protocol's pre-set rules, allow us to determine if medical "hope" is backed by statistical "certainty."
Practical Implications: The "So What?" of Protocol Literacy
For the non-specialist, protocol literacy is the ultimate tool for distinguishing "medical hype" from genuine "medical hope." When a headline screams about a new "cure," understanding the trial's design allows you to evaluate the strength of the evidence. Was the sample size large enough to be meaningful, or was it a "pilot" study involving only a dozen people?
When reading about a new clinical study, use this checklist to evaluate its strength:
Sample Size: Did the study include enough participants to ensure the results aren't just a fluke?
Duration: Did the study last long enough to reflect the chronic nature of the condition or to catch long-term side effects?
Funding Sources: Who paid for the study? Transparency in funding helps identify potential conflicts of interest.
Conclusion: The Future of the Blueprint
The clinical trial protocol remains the bedrock of modern medicine, a testament to the human requirement for order in the face of biological complexity. As we look forward, we are seeing the rise of "Adaptive Trial Designs"—innovative protocols that allow for pre-planned modifications based on interim data, potentially speeding up the delivery of life-saving drugs without sacrificing rigor (FDA, 2022).
There is a profound sense of wonder in the realization that every life-saving pill in a medicine cabinet began as a series of carefully typed pages. It is the ultimate expression of human ingenuity: the ability to map the journey from a laboratory molecule to a human treatment, ensuring that the path to health is paved with evidence, ethics, and certainty.

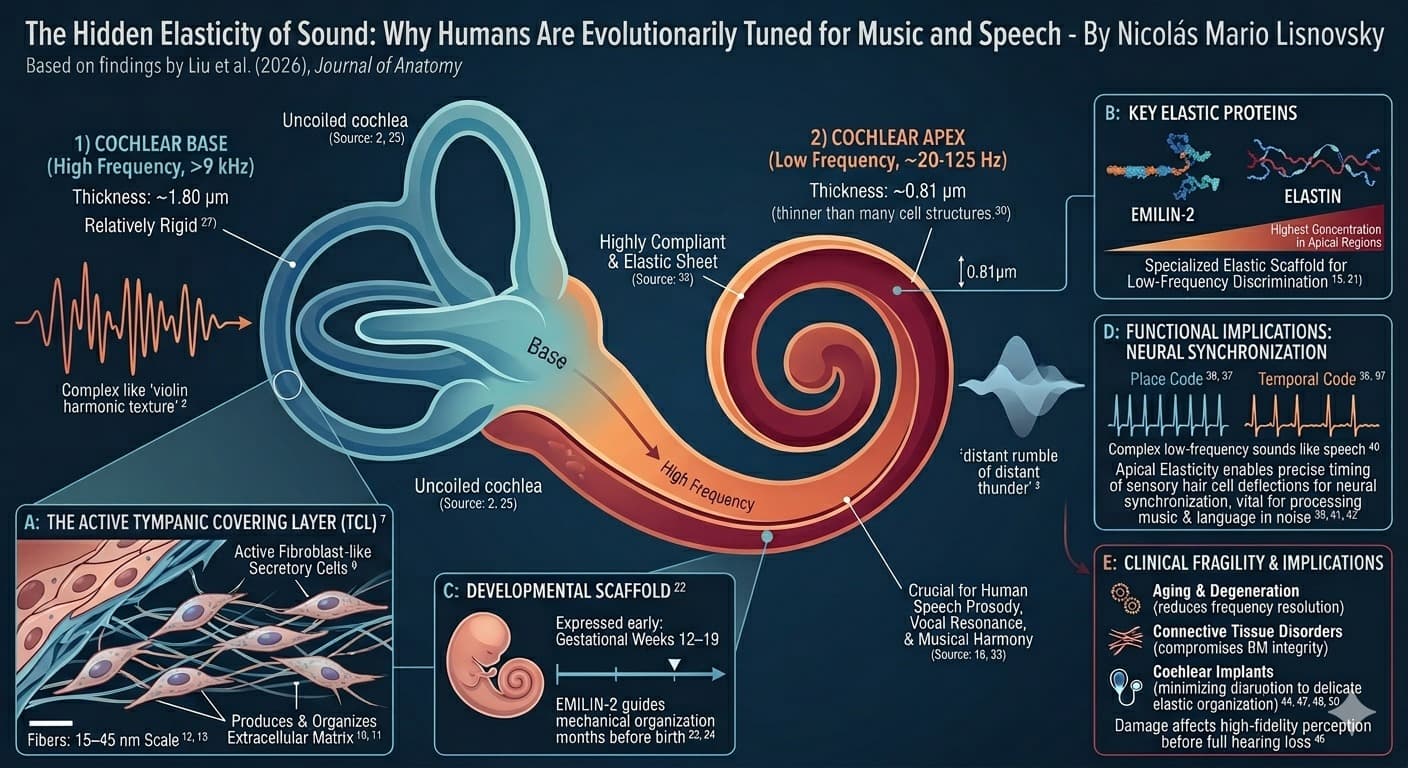
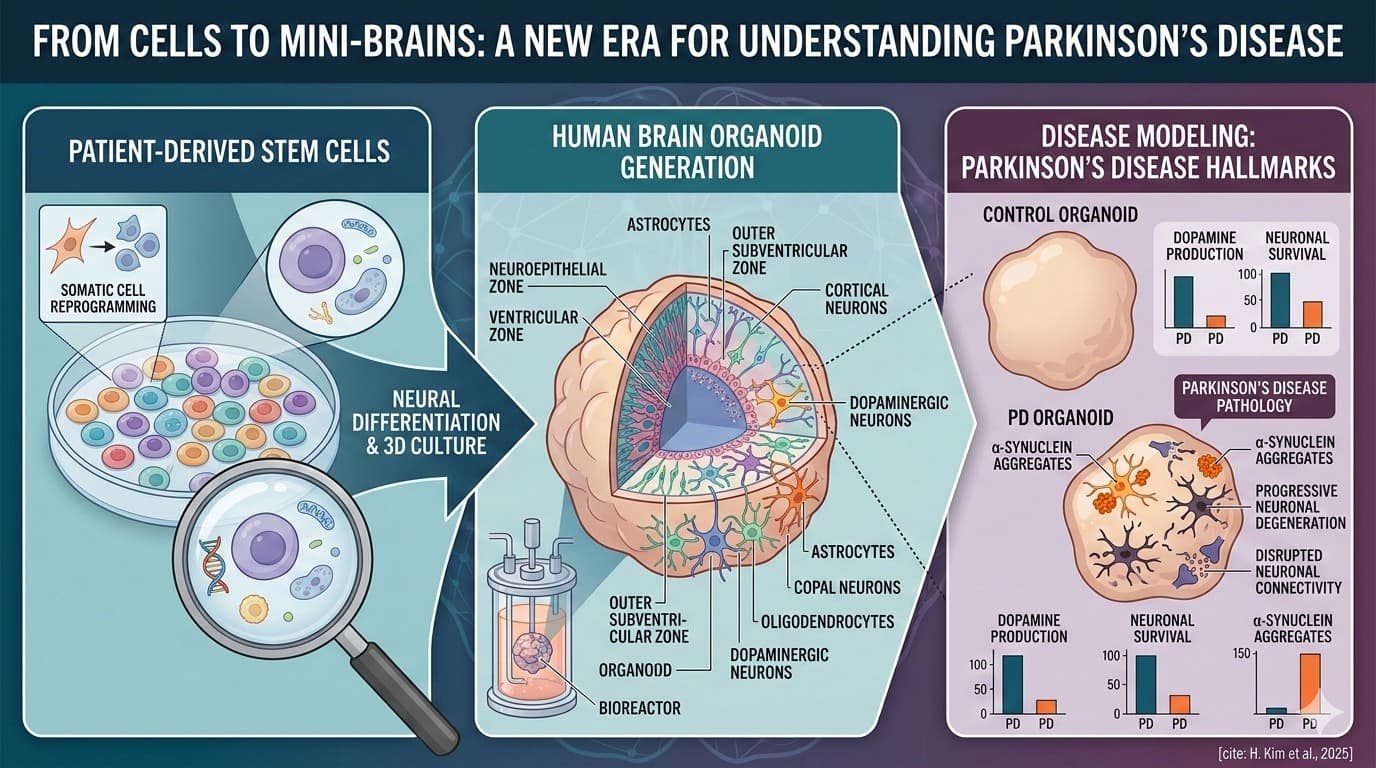
Discussion